The discovery of anti-Perforin-2 effectors and elucidation of their molecular mechanisms is an area of significant interest to our lab.
The gene encoding Perforin-2, Mpeg1, has existed for hundreds of millions of years and is present in nearly all animal species; including those within the phylum Porifera (sponges) which evolved 500 million years ago. Thus, Perforin-2 has been protecting even the simplest of animal species from infectious diseases since before dinosaurs walked the planet. The evolutionary longevity of Perforin-2 is a testament to its pivotal role within cell-mediated immunity. However, this has also allowed pathogens ample time to evolve mechanisms that inhibit and subvert Perforin-2’s bactericidal activity.
One of our collaborators, Prof. Nikki Traylor-Knowles at the University of Miami Rosenstiel School of Marine and Atmospheric Science, is studying the evolution and expression of Perforin-2 in corals. Their study has been accepted as part of a special emphasis article collection on Perforins and Cholesterol-Dependent Cytolysins in Immunity and Pathogenesis. Several other collaborators from the University of Miami have also contributed manuscripts to this collection.
In theory bacterial pathogens have likely evolved numerous strategies to survive Perforin-2. Some may suppress the expression of Perforin-2. Others may escape the early endo-phagosome before Perforin-2 is fully deployed. Some may prevent phagosome acidification to inhibit the acid-dependent pre-pore to pore transition. Still others may deploy effectors to prevent the intracellular trafficking of Perforin-2, see Cif below for a specific example, or its maturation. Hypothetically, any step along the Perforin-2 pathway –from PAMP signaling and initial expression to the final step of pore formation– is a potential target for pathogen counter defense. In this respect anti-Perforin-2 effectors can be thought of as molecular probes that help map the Perforin-2 dependent killing pathway through elucidation of an effector’s mechanism of action. Thus, the discovery of anti-Perforin-2 effectors is a major area of interest to the Munson lab.
Cif (cell cycle inhibiting factor)
The short, 38-39 residues, cytosolic tail of Perforin-2 contains three conserved lysine residues that we have shown are critical for the intracellular trafficking of Perforin-2 to endo-phagosomes (McCormack et al. eLife 2015). We have also shown that PAMPs such as LPS trigger the monoubiquitylation, or poly-monoubiquitylation, of Perforin-2 and that this covalent modification initiates a cascade of events that mobilize Perforin-2 to its final destination. Thus, the addition of ubiquitin to one or more of the conserved lysine residues can be thought of as a delivery address that is read and acted upon by the protein trafficking and sorting machinery of host cells. We have further identified the E3 ligase responsible for attaching ubiquitin to Perforin-2’s lysine residues. The ligase is a multi-component complex of proteins known as a cullin-RING E3 ubiquitin ligase (CRL). At its core lies the cullin CUL1 which acts as the scaffold for CRL assembly. This particular complex also contains SCF-betaTrcP which is involved in substrate recognition; in this case the cytosolic tail of Perforin-2. Crucially cullin neddylation –the covalent ligation of the ubiquitin-like peptide NEDD8 to a substrate– drives ubiquitylation of the CRL substrate.
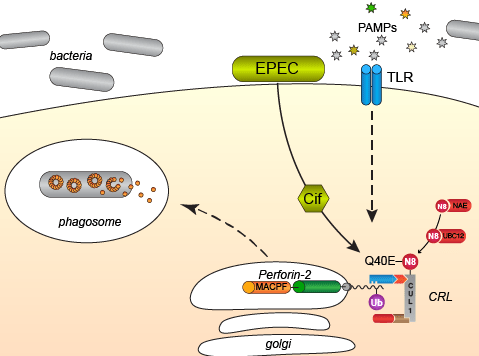
Cif’s are bacterial effector proteins that are injected into the cytosol of host cells by bacterial Type III protein secretion systems. Once inside a host cell Cif’s inactivate CRLs by enzymatic modification, specifically deamidation, of NEDD8. Thus, Cif’s block all CRL-dependent ubiquitylation within host cells. As expected, we have shown that ubiquitylation of Perforin-2 and Perforin-2 -dependent killing is blocked by bacteria that express Cif but not mutants in which cif has been deleted. We have also shown that Cif+ pathogens are more virulent in vivo than cif mutants (McCormack et al. eLife 2015, Figure 8). Crucially, Cif does not confer a selective advantage in Perforin-2 knockout animals (McCormack et al. eLife 2015, Figure 9). In other words, when Perforin-2 –the crucial target/substrate molecule– is absent, the pathogenic benefits of Cif are nullified. Thus, host cells ubiquitylate and deploy Perforin-2 to defend themselves against microbes. As a countermeasure some pathogens deploy Cif’s to block the intracellular trafficking of Perforin-2 to endo-phagosomes.
Cif’s are likely just one example of numerous strategies that bacteria deploy to inactivate or otherwise avoid the bactericidal activity of Perforin-2.
